

TLDR and/or Not Wolbachia Inclined?
We only name that which is a part of us…
… our children, our pets, our creations. Only our deepest passions get named. Thus, can anyone not be opinionated about nomenclature? I used to think people who argued about nomenclature were idiots, and I was right. Now I think people who don’t argue about nomenclature are idiots, and I am still right!
Catch yourself the next time you think another person’s kid’s names are dumb, you think this because your background dictates what names make sense to YOU; names are deeply selfish things!
Let us take ourselves both infinitely seriously… and not too seriously. After all, no one’s ridiculous names will last too long; not even mine…
“All that you touch; All that you see; All that you taste; All you feel. All that you love; All that you hate; All you distrust; All you save. All that you give; All that you deal; All that you buy, beg, borrow or steal. All you create; All you destroy; All that you do; All that you say. All that you eat; And everyone you meet; All that you slight; And everyone you fight. All that is now; All that is gone; All that’s to come and everything under the sun is in tune; but the sun is eclipsed by the moon.” – Pink Floyd
 I was asked by a colleague to give my thoughts on this preprint. I reached out to Dr. Newton and provided her and Dr. Lindsey the last word, albeit in <small> print. 😉
I was asked by a colleague to give my thoughts on this preprint. I reached out to Dr. Newton and provided her and Dr. Lindsey the last word, albeit in <small> print. 😉
I am commenting on this paper because it is entirely about the CI loci. According to my reading (Full Disclosure* I am reading into all the subtleties and politics of this paper, past just what is written in words), this pre-print postulates two main premises, both of which have some flaws:
1. Differential transcript abundance between A and B ORFs means they are not an operon and thus they are not a toxin-antidote.
While we do not think that the A&B ORFs represent a TA-operon, the operon hypothesis does not preclude their function as a TA system. In the manuscript, we do not use our analysis of operon-like behaviors to infer whether or not these genes function as a TA system. Instead, we point to the interesting and divergent pattern of gene expression for the two loci, suggesting a more complex regulation than a simple operon hypothesis would explain.
2. The deubiquitylase (DUB) domain is not the root cause of CI because other biochemical domains might exist within CidB. Furthermore, other CI systems relying on alternate biochemistries undoubtedly exist.
This is not what we imply. Indeed, the DUB-related CI function is convincing in wPip and we do not imply that other domains within that allele are instead responsible for the phenotype. They may be important for other functions (binding, localization, etc.), but our domain searches were performed with the intent of discovering the diversity present across more divergent alleles.
With respect to point 1. The first half of the argument is whether or not these genes constitute an operon. Here, I don’t disagree with their data, only the interpretation. In the end, operon or not, this is a nuanced semantic argument about complex bacterial regulation of genes; the semantics don’t really change the functional implications of any group’s conclusions or data. All of us agree that:
a) the two genes are genetically linked and always found together (synteny).
b) the two genes are functionally linked, bind one another, and are both involved with CI somehow.
c) the two genes are polycistronically transcribed.
Under these premises the genes already meet 3 important characterizations of bacterial “operons.” However, there is disagreement about the extent of polycistronic transcription, how functionally relevant this is, and a final point d) Co-regulation, which is at the heart of operon mechanics.
We disagree on whether or not this is semantics. Regulation of these genes in the host is an important consideration, especially for Wolbachia-based vector control programs to be implemented effectively. We do not agree with c). We conclude the opposite from our transcriptomic and qPCR data.
The authors conclude and verify previous reports 1 that polycistronic transcription does occur in CidA/BwMel, but the authors argue that this is just accidental leaky read through of an RNA polymerase…some kind of inefficient termination. I believe this is NOT an accident. I believe this is, in fact, how it works! Imagine taking a hypothetical bacterial operon of any function, say the lac operon: lacZ, Y, A:
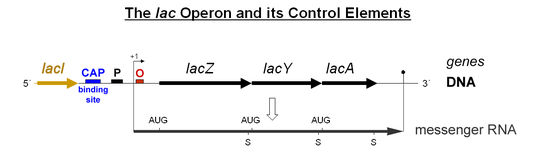 Now imagine that a cellular environment changes and a bacterium no longer wants to have equal stoichiometry of Z, Y, A. This would be very easy to engineer into the operon’s transcription, just add a cryptic terminator in gene Y/A or an RNA hairpin/Rho binding structure that makes the polymerase fall off and/or finish the transcript less efficiently; even easier, just make the last gene, A, really really long… like CidB… Such adaptations would make transcripts Z and Y more efficiently transcribed and thus more abundant than a full-length polycstronic transcript. The bacteria could use this as a technique to tweak transcript stoichiometry. Is a tweaked lac operon still a lac “operon?” I would say, “Yes, it is.” Thus, in my opinion, simple differences in transcript abundance says nothing about whether the loci are, or are not an “operon.”
Now imagine that a cellular environment changes and a bacterium no longer wants to have equal stoichiometry of Z, Y, A. This would be very easy to engineer into the operon’s transcription, just add a cryptic terminator in gene Y/A or an RNA hairpin/Rho binding structure that makes the polymerase fall off and/or finish the transcript less efficiently; even easier, just make the last gene, A, really really long… like CidB… Such adaptations would make transcripts Z and Y more efficiently transcribed and thus more abundant than a full-length polycstronic transcript. The bacteria could use this as a technique to tweak transcript stoichiometry. Is a tweaked lac operon still a lac “operon?” I would say, “Yes, it is.” Thus, in my opinion, simple differences in transcript abundance says nothing about whether the loci are, or are not an “operon.”
This explanation laid out here would explain how one would find decreasing numbers of transcripts of genes more 3’ within the region, but it does not explain how you would get higher expression of more 3’ genes (such as the B-gene in our system), which we show does occur at several time points in host development – ex. early embryogenesis, and late larval stages. This point is especially critical in our assertion that this is not an operon-like pattern of transcription. As we assert above, the mechanism behind this complex regulation needs investigating as it is not easily explained and it is why we highlight the pattern of expression observed.
The final criteria for an “operon” would be co-regulation by a single promoter and terminator. The authors here imply that there must be a cryptic promoter within cidA which differentially regulates the second gene cidB, and that the two genes are regulated by alternative transcription factors, yet the authors do absolutely no experiments to test this or validate this in any way. Furthermore, I would argue exactly the opposite has already been verified in my previous publication.2 We take both Cid and Cin operons and put them into an arabinose promoted plasmid with the full length wildtype operon sequences and successfully drive expression of BOTH genes from the single ara promoter in pBad. This expression is arabinose dependent. If there was a cryptic promoter, one would expect that the B protein would just continuously express in the absence of arabinose, but this does not happen. Thus, in E.coli, the genes are “operon enough” to work as an operon under a single regulatory element (the ara inducible promoter).
We do not discuss or mention [cryptic promoters] at all. Indeed, a paucity of transcription factors exist in endosymbiont genomes broadly and in Wolbachia few have been characterized [but see this reference]
To be fair, no experiments have yet been done to support the hypothetical cryptic terminator in gene A or an RNA hairpin/Rho binding structure.
It is important to remember that this is a heterologous system. While useful in many respects, it may not be appropriate for looking at native transcription. For example, your hypothesized transcription factor that would drive the B-gene may not be present. Indeed, we have no sense of how the Wolbachia promoters would be recognized by the E.coli machinery (that is presumably why you drove expression with the arabinose promoter).
The second point of their first argument is that because the genes are not an “operon” and have differential expression then they cannot be a toxin-antidote system. This is incorrect. Most TA systems actually have encoded genes that auto-regulate transcription of their own ORFs. What if CidA binds and sits on the CidB genomic ORF limiting transcription of CidB? Furthermore, a TA system would actually want more antidote transcript than toxin! So the transcriptomics, in my opinion, don’t discredit the TA argument.
Again, we do not argue that this is not a TA system based off our operon findings. These are separate considerations.
We agree [that] the operon hypothesis and expression levels are different from the TA hypothesis. What is critical to consider, however, is that both A and B are required for CI induction in the fly. Additionally, CidA transgenic expression does not rescue the CI nor the CI-like defects induced upon CidB expression. This implies that cidA/B, cifA/B are not classical TA systems.
Rescue will only be figured out, and TA system disproven or proven, when one great scientific hero can demonstrate molecular rescue of the transgenic CI system by a single and or complex of ORFs. Then, and only then, will the TA system be disproven or proven. This person, maybe myself, will have my undying adulation and praise and a new Nature paper.
Now to Point 2. The deubiquitylase (DUB) domain is not the root cause of CI because other biochemical domains might exist within CidB and other CI systems relying on alternate biochemistries undoubtedly exist. This is now the sole perpetual thesis of every paper about CI, on which, Dr. Bordenstein is an author. Discussion of this point inevitably must go through a nuanced and political debate about nomenclature, but hey, I’m all about honesty, and the debate needs to happen. If the DUB domain is not important for CI, Bordenstein is able to justify the Cif nomenclature, CI inducing factor, which is entirely stupid because it completely ignores all my and Dr. Ronau’s biochemistry.2 We CLEARLY demonstrated that the DUB domain is essential for transgenic CI, in those alleles.2 That is why we call them Cid, CI inducing Deubiquitylase. We proved that both CidBwPip and CidBwMel (Seth’s Cif) are BOTH DUBs! We also point out clearly in our paper that we believe there are alternate biochemistries capable of inducing CI (not just the DUB). We highlight these genes, name them Cin, for CI inducing nuclease, and validate their catalytic triad, DEK, in yeast. Thus, in strains like wNo, wRi, and others, that do not have functional Cid deubiquitylase operons, we argue that in lieu, these nucleases probably induce CI. The evidence for this, published so far, is that Medea elements seem to rely on the same nuclease biochemistry3 and that in yeast, toxicity of CinB is alleviated by mutagenesis of the catalytic nuclease triad.2
The key word here is “alleles”. Being evolutionary biologists, we are very much interested in the diversity of these genes across Wolbachia. Your DUB work in the wPip strain clearly shows the importance of that domain/residue in that allele, but our work aimed to generate additional hypotheses regarding what may be going on in the more divergent alleles.
As you can see from our analysis, these residues are not conserved across Wolbachia. And again, we’re interested in conservation/divergence across strains.
Indeed – it would be really fun to determine support for [the Nuclease CI Induction] hypothesis – and relates to our discussion of the possible routes of CI in the non-DUB containing alleles.
Nomenclature of these genes can be easy and informative! In my view, there is a simple dichotomy of biochemistries that seem to be involved with CI induction in Wolbachia. The Cid enzymes rely on a DUB and the nuclease paralogs/orthologs rely on a nuclease. ALL known orthologs of “Cif” genes, and I have looked at these more than any other person on the entire planet, are either nucleases, DUBs, or have both functional catalytic triads. This is precisely why I differentiate nomenclature based on this key biochemical Dichotomy. When using “Cif”, one has no fucking clue if one is talking about a DUB, a nuclease, or both. It is completely uninformative, unhelpful, and confounding.
I think we have differing opinions on what constitutes “easy” nomenclature. Additionally, as the biochemistry has not been worked out for these other homologs, the conservative approach is to consider them Cif genes for now.

Dr. Newton knows how much I hate strawmen… Touché!
Finally, now to their actual point about the biochemical roots of CI. We demonstrated that the DUB is essential for CI (in those alleles). It is hypothetically possible that the root cause of induction might be a different biochemistry. But the authors don’t back this up with any data or experiments. They just use a “junk-in junk-out” prediction program like HHpred to guess about what other biochemistries could possibly exist. These computer programs are designed to give ideas that should be tested, not to make conclusions about biochemistry. Furthermore, this type of argument can go on ad infinitum, “Sure the DUB might be important, but what about this other amino acid! No one has tested that one! What about all the other possibilities you haven’t tested!” This is a classic straw man argument! If the authors have an alternative hypothesis for CI induction by any means other than a DUB or nuclease, which we postulated as the roots of CI, the burden of proof is on these authors to biochemically validate this. Providing a list of other possibilities could be helpful, but it can also muddle the mixture in the same way that the “ANK genes inducing CI rumor” did for 10 years. What about these ANK genes, these operons can’t be involved with CI because look at all these ANK genes sitting around!? Well, there was never actually any reliable data showing those ANK genes were important to CI in any way… There is strong data showing the DUB and nuclease triads are important!
We did our best to be very clear that these results are hypotheses and should not be taken at face value. Our point was not that every amino acid should be tested but that these different homologs, which lack DUB domains, should be investigated to determine if they induce CI and if so, by what mechanism. We do offer some directions in the preprint, but are very careful to not focus on them, as we agree: we don’t need an ANK re-do.
 In conclusion:
In conclusion:
a) We all agree there must exist alternative biochemistries involved with CI induction, besides the DUB. Sullivan at the last meeting said, “CI must be incredibly easy to evolve… You just have to mess up chromosomes…” There must also exist an alternative or possibly convergent biochemistry in Cardinium CI; But the DUB is undeniably important, and probably is in fact, the root of CI for Cid-type enzyme systems. Yep. Agreed. The Cid system likely evolved from a common nuclease CI ancestor into a DUB biochemistry; we hypothesize this because the missing links between the two divergent paralogs, enzymes that have both nuclease and DUB triads, exist in the database right now and the Cid enzymes exhibit a devolved nuclease skeleton secondary structure with the loss of the active triad2…
b) They don’t have any data arguing against a classic toxin-antidote system. Their essential premise is wrong, that differential transcript abundance means the two loci are not an operon, and thus they are not a toxin-antidote. If they want to definitively conclude against the TA hypothesis (which has evidence to support it in yeast and comparative genomics,2 they must find the alternative rescue factor, maybe they have? There is also a possibility that the TA hypothesis is just more complex; a third factor that mediates or modulates the interaction of A with B proteins. This could be a key host protein/modification or a Wolbachia protein. The only real data against the TA hypothesis is negative data: that the transgenic CidA constructs in both papers would not rescue the transgenic CI.2,4 But no one has even validated they get the protein CidA to the right place, at the right time, with the right modification or folding status, to actively rescue, thus no one can use this data to “disprove” or “conclude” anything about the TA hypothesis because no one has done the controls to validate that these experiments are actually testing that hypothesis correctly. Whatever the rescue factor is, it needs to be expressed properly, folded properly, modified properly, and pre-loaded and localized properly into the embryo before the first 20 minutes of embryogenesis. Maybe our dual groups did this properly, maybe we didn’t? I welcome the day when some great hero figures rescue out. I want this to happen, and I am trying my best to make it happen! We also look forward to the discovery of the rescue, whatever it may be.
To reiterate, this is not our premise. We can make it clearer in a revision, that these are separate sets of data, and we really only look at the operon hypothesis. Any conclusions about the TA system come from the necessity of both ORFs for CI in the host.
We agree that the transgenic experiments have significant drawbacks – as do heterologous systems. But negative data are data. The fact that CI can be induced but the rescue can’t, should cause you some pause.
c) The nomenclature mess is boiling over. Like it or not it seems like “Cif” refers to all orthologs, paralogs, and analogs from any and all Wolbachia Underneath this is a dichotomy of nuclease vs DUB functionalities which is defined by Cid vs Cin nomenclatures. Cif users want to hold out and wait to definitively name the genes at the time they feel the root biochemistry has been clearly defined. Cid and Cin users feel the root biochemistry has already been defined! The field, future publications, and time will decide what is the best and easiest system.
There is evidence that your suggested nomenclature makes things more difficult to comprehend – that some folks think that the cid loci are not homologous to the cif loci.
This preprint has an opportunity to make things simple. I encourage the authors to think about this: If as a field we make nomenclature of these loci so abysmally difficult to understand that no one can read a paper without knowing the intricate details of Type I-IV…V… VI CI systems, each with Modules A, B, C, D, E, F, G all of which would need to be cross checked with a table to figure out what they hell these modules actually biochemically do ect… we will make our own papers incomprehensible. I would encourage the authors to define gene nomenclature based on proven biochemical functions only! Do not go the way of Cif! It leads down a dark road of incomprehensible jargon; I, II, III, IV, V, VI; modules A, B, C, D, E, F G…!
It seems as though there are some misunderstandings (ex.TA function directly related to operon behavior, the role of DUBs within vs. across alleles). We are happy to re-word such that these misunderstandings are mitigated, and appreciate your attention to the manuscript. If there are cool regulatory mechanisms that result in these drastic and sometimes inverted A/B ratios we look forward to learning about them. For now, we follow the data. No doubt, the DUB is critical in wPip, and we are fascinated by the rapid and divergent evolution of these genes across Wolbachia strains.
References:
1 Beckmann, J. F. & Fallon, A. M. Detection of the Wolbachia protein WPIP0282 in mosquito spermathecae: implications for cytoplasmic incompatibility. Insect biochemistry and molecular biology 43, 867-878, doi:10.1016/j.ibmb.2013.07.002 (2013).
2 John F. Beckmann, J. A. R., Mark Hochstrasser. A Wolbachia deubiquitylating enzyme induces cytoplasmic incompatibility. Nature Microbiology 2 (2017).
3 Lorenzen, M. D. et al. The maternal-effect, selfish genetic element Medea is associated with a composite Tc1 transposon. Proceedings of the National Academy of Sciences of the United States of America 105, 10085-10089, doi:10.1073/pnas.0800444105 (2008).
4 Daniel P. LePage, J. A. M., Sarah R. Bordenstein, Jungmin On, Jessamyn I. Perlmutter, J. Dylan Shropshire, Emily M. Layton, Lisa J. Funkhouse-Jones, John F. Beckmann, Seth R. Bordenstein. Prophage WO genes recapitulate and enhance Wolbachia-induced cytoplasmic incompatibility. Nature 2 (2017).